Caught between the American and French Revolution: http://youtu.be/nMU8aa1IHfQ
The Trinity in the Fourth Century: https://youtu.be/pNI2HzN5EMQ
Caught between the American and French Revolution: http://youtu.be/nMU8aa1IHfQ
The Trinity in the Fourth Century: https://youtu.be/pNI2HzN5EMQ
Michelson and Morley thought we were going through a static ether
Light came back at same time so said no ether
Dynamic ether
Disproved a static ether
Maxwells equation needs a medium for waves to move
Ether, medium for propagation of light, is electron-positron pair {D.L. Hotson says the same in his negative energy solution to Dirac’s equation paper}
Electron positron (like a dipole) annihilate -> release energy and go back into ether -> speed of propagation of electron-positron ether = speed of light
Claim 1. All wave propagation requires a medium
Quantum field = space time = Higgs field = ether
E-p lose energy when go back into ether (lower end state for complexes/ compounds) -> lose most mass energy
Stiffness = inverses permeability, must be high; can still be elastic
Density is very low of e-p
The more solid the faster the speed of light (diamond vel >> air)
Any particle antiparticle (p-pbar) will annihilate back to the ether
Photon = light, when you stand in a water wave it pushes you like a circle, wave is propagating towards the shore so it’s massless, the water has mass
When the water hits the shore it breaks (then water is moving) – eye/ camera as a shore so when wave crashes into it its now a particle = wave particle duality (so what is the mechanism that then gives it mass?)
Wave is a perturbation
Light bulb is vibrating the medium which propagates the wave
Speed of propagation of the medium doesn’t depend on if you’re moving with respect to it. Might see a Doppler shift in one direction.
(Why are the electron positron pairs so much smaller than the electron?) = electric fields
e-p ether reacts to charge coming in and flips orientation towards
Tesla, Einstein, and Maxwell all invoke the ether
Thoughts
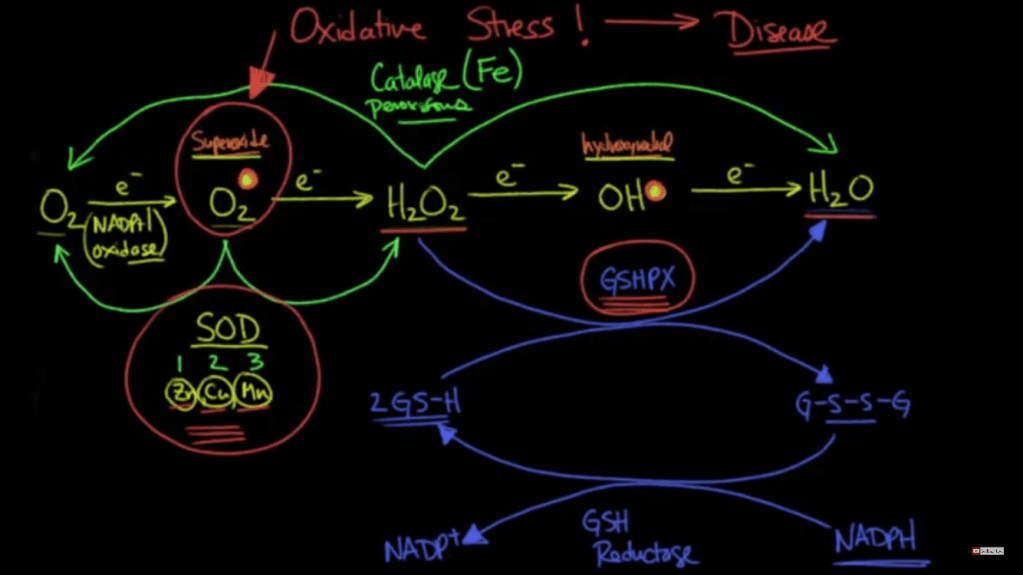
The challenge- answer how oxygen gets to complex IV in the mitochondria to make water. Logically diffusion doesn’t make sense as the mechanism for enough oxygen to be made available to the 9000rpm ATPase found in mitochondria. Especially if a 70kg man only intakes ~250ml~0.016moles~9.41E21 atoms of oxygen each minute. While this doesn’t fully answer that question, I think it’s as very plausible mechanism. There would have to be melanin in the mitochondria matrix too.
This makes me wonder how the CO2 leaves the cell.
Notes
Normal cells make lactate under anaerobic conditions
Cancer and embryonic cells preferentially send glucose to make lactate even under aerobic conditions = aerobic glycolysis (Warburg effect)
Highly proliferative cells need to produce excess lipid, nucleotide, and amino acids for the creation of new biomass. Excess glucose is diverted through the pentose phosphate shunt (PPS) to create nucleotides. Fatty acids are critical for new membrane production and are synthesized from citrate in the cytosol through the action of ATP-citrate lyase (ACL) to generate acetyl-CoA.
glucose is only source of biomass, and not energy, for the latter our body is able to take it from the water through dissociation and back together of water molecule.
Hemoglobin has the ability to irreversibly dissociate in water molecule, as does another molecule that is almost identical to it: chlorophyll. In this way the oxygen that enters the blood and is carried mainly by the Hb not only comes from the pulmonary alveoli but hemoglobin itself.
The main function of the oxygen in the lungs and blood is to facilitate the transport of
CO2, the allosteric modification that produces the oxygen in the hemoglobin molecule is important in the dynamics of CO2, is an observed fact that hypercapnia, hypoxia worsens.
melanin has the amazing ability to dissociate and re-form the water molecule, using the entire electromagnetic spectrum, as melanin is able to absorb any type of energy, is the substance for something more known dark, humans thus dissociate and re-form the water molecule both day and night.
If the oxygen was important, we would be before a tightly regulated process, which is much more compatible with life than a system that is based on the laws of simple diffusion
when water molecule is cleaved, the photon energy is transformed into free chemical energy, which is transported by the molecular hydrogen, which, in turn, is not combined with water. In the case of hemoglobin, color allows us to think that is absorbing visible electromagnetic radiation mainly close to purple, as this reflecting the waves near the red, say between 650 and 700 nanometers, therefore our senses perceive erythrocytes red.
for a moment put into the equation the property intrinsic of hemoglobin to dissociate the water molecule, things change, because if the erythrocyte is producing oxygen continuously, then O2 levels do not decrease until the CO2 increases, since the carbon dioxide decreases oxygen levels at least two reasons: causes an allosteric change in the hemoglobin molecule which decreases its affinity for O2 and also its efficiency to dissociate the water molecule. The properties of water are also changed by higher concentrations of the CO2 molecule.
chlorophyll, which expellee oxygen to the atmosphere, hemoglobin expels oxygen to their immediate environment, and part of it is taken up by the prosthetic group of hemoglobin: iron.
Water can dissolve thousands of times more CO2 than air, so the amniotic fluid with carbon dioxide form bicarbonate, and does so very quickly thanks to an enzyme called carbonic anhydrase. Thus the fetus has no need to breathe inside the uterus. After birth, the CO2 be ejected into the atmosphere, so the respiratory rate rises abruptly to 30-60 breaths per minute during the first year. This change in respiratory rate is needed to compensate the rate at which CO2 is diluted in the air.
Albumin has a strong negative charge, but there is little correlation between the charge of the compound and the degree of binding to albumin. Most is recovered back into the circulation by lymphatic drainage. Albumin lost into the intestinal tract (1 g each 24 hours) is digested releasing amino acids and peptides which are reabsorbed. Albumin synthesis in humans takes place only in the liver. Albumin is not stored but is secreted into the portal circulation as soon as it is manufactured, about 12-25 g of albumin per day. The synthetic pathway is common to eukaryotes and is also used for synthesis of other proteins.
intrinsic property of hemoglobin irreversibly dissociate the water molecule
melanin as photonic energy transducer in free chemical energy are consistent with the findings that hardly pass oxygen blood-brain barrier. For one oxygen needs are covered by the dissociation of the water molecule, and energy needs are largely solved by molecular hydrogen, which when not combined with water can reach the remotest area of the cell, it follows the laws of simple diffusion, conducting at least two important functions: delivery of energy and also as an antioxidant, since hydrogen is the best.
Thoughts
They corroborate the theory that Fe+2 will make it through the VGCCs. Many used to talk about trying calcium channel blockers to deal with nnEMF effects. They cause my blood pressure to spike and the need to go on oxygen so I won’t be trying this anytime soon. Maybe a low dose natural CCB? Really I would rather get the Fe out of the cell with TTFD. Especially because the paper says this only occurs in cells that express many LVDCCs.
If the CCBs are reducing Fe levels in the cardiac myocytes then what is all that Fe doing in the blood and elsewhere? Still need to address getting it out of the body.
The ability for CCB to block Fe+2 uptake depends on the frequency of the membrane. This is very interesting especially given Fe uptake doesn’t seem to depend on voltage or density.
“L type means it’s long lasting. They are responsible for the excitation-contraction coupling of skeletal, smooth, cardiac muscle, and for aldosterone secretion in endocrine cells of the adrenal cortex. They are also found in neurons, and with the help of L-type calcium channels in endocrine cells, they regulate neurohormones and neurotransmitters. They have also been seen to play a role in gene expression, mRNA stability, neuronal survival, ischemic-induced axonal injury, synaptic efficacy, and both activation and deactivation of other ion channels.”
“The most predominant way of autoinhibition of L-type calcium channels is with the Ca+2/Cam complex. As the pore opens and causes an influx of Calcium, calcium binds to calmodulin and then interacts with the loop that connects the adjacent EF-hand motifs {bind Ca+2} and causes a conformational change in the EF-hand motif so it interacts with the pore to cause quick inhibition in the channel. It is still debated on where and how the pore and EF-hand interact. Hydrophobic pockets in the Ca+2/Cam complex will also bind to three sections of the IQ domain known as the “aromatic anchors”. The CA+2/Cam complex has a high affinity towards L-type calcium channels, allowing it to get blocked even when there are low amounts of calcium present in the cell. The pore eventually closes as the cell repolarizes and causes a conformational change in the channel to put it in the closed conformation.”
https://en.m.wikipedia.org/wiki/L-type_calcium_channel
Given the organs that express the most LVDCCs, Morleys thoughts about Fe buildup causing what we term diseases is along the right path. Though it’s interesting that liver cells don’t express functional ones. Maybe Fe isn’t the cause of liver and liver related issues.
NTBI is non-transferrin bound iron – this is unbound iron.
Highlights
hypothesize that in iron-overload disorders, iron accumulation in the heart depends on ferrous iron (Fe2+) permeation through the L-type voltage-dependent Ca2+ channel (LVDCC), a promiscuous divalent cation transporter. results indicate that cardiac LVDCCs are key transporters of iron into cardiomyocytes under iron-overloaded conditions, and potentially represent a new therapeutic target to reduce the cardiovascular burden from iron overload.
The prevalence of primary (hereditary) hemochromatosis and sec- ondary iron overload (hemosiderosis) is reaching epidemic levels worldwide1–5. Primary hemochromatosis is the most common genetic disorder in white individuals of European ancestry, with an allele fre- quency >10% (refs. 2,6). Iron overload leads to excessive iron deposi- tion in a wide variety of tissues, including the heart and endocrine tissues7–9. Iron-overload cardiomyopathy is the main determinant of survival in patients with secondary iron overload10–12. Although liver dysfunction and cirrhosis are the main causes of death in patients with primary hemochromatosis3,13, iron-induced cardiac dysfunction is also a leading cause of morbidity and mortality in these patients13–16. Elevated cardiac iron causes marked diastolic dysfunction, increased propensity for arrhythmias and late-stage dilated cardiomyopa- thy12,14–19. In addition to cardiac dysfunction, iron-overloaded patients routinely suffer from a range of endocrinopathies, including diabetes mellitus and anterior pituitary dysfunction3,8,20,21. The cur- rent mainstays of therapy for excessive iron deposition in patients with primary and secondary hemochromatosis are phlebotomy and iron chelation, respectively, which are designed to promote whole-body iron removal. Unfortunately, patients with primary hemochromatosis are often treated only after iron overload becomes advanced13,15,22, and chelation therapy is cumbersome and associated with toxic side effects, with only a limited impact on clinical outcome in patients suf- fering from secondary iron overload.
Generally, cellular iron uptake into cells is tightly regulated and occurs through a combination of transferrin-dependent and trans- ferrin-independent (non-transferrin-bound iron, or NTBI) path- ways3,27–29. Under conditions of iron overload, the trans- ferrin-dependent system becomes inhibited and excessive iron accu- mulation occurs predominantly through the NTBI pathway3,27–31. Iron uptake through the NTBI pathway activates a positive feedback system whereby the uptake of iron is further enhanced27,28,30. Several metal transport systems such as LVDCCs, which show promiscuous permeation by divalent metal ions32,33, and the divalent metal trans- porter-1 (DCT-1/DMT-1) can contribute to NTBI uptake in heart and other tissues. The DCT-1/DMT-1 system is expressed weakly in the heart and, like the transferrin system, its expression is negatively regulated by iron levels29,31,34, suggesting a limited role in NTBI uptake in the heart. In contrast, cardiac sarcolemmal LVDCCs are expected to be especially important for the excessive NTBI uptake under conditions of iron overload for several reasons. First, NTBI uptake involves the transport of the reduced form of iron (Fe2+), which is the iron species permeating LVDCCs in cardiomy- ocytes28,30,32,33 and Langendorff hearts32. Second, as with NTBI uptake, LVDCC currents can be increased when the concentration of Fe2+ is elevated32. Third, a common feature of cells susceptible to iron overload, such as cardiac myocytes, pancreatic β-cells and anterior pituitary cells, is the large number and activity of LVDCC
specific analysis of the collagen volume fraction, using picrosirius red–stained sections, revealed a sixfold increase in the iron + vehicle group (0.53 ± 0.18 for placebo + vehicle versus 3.4 ± 1.2% for iron + vehicle; n = 4; P < 0.01). Moreover, in mice injected with iron and treated with verapamil, the colla- gen volume fraction was markedly reduced
significant increases (P < 0.01) in cardiomyocyte apoptosis in mice injected with iron (Fig. 2f), suggesting that the increased fibrosis in these mice resulted in part from increased myocyte loss. As with the functional changes, treatment with CCBs reduced the composite index of myocardial damage and inflammation and the degree of apoptosis by similar amounts
CCBs selectively reduced the iron deposition in myocytes without affecting extramyocyte iron accu- mulation (Fig. 2c,d,g). Analytical electron microscopy coupled with X-ray microscopic analysis confirmed iron accumulation in cardiomyocytes and showed that iron is deposited throughout the sarcoplasm, particularly in the perinuclear and subsarcolemmal regions as iron-containing electron dense bodies
hepatic iron levels were unaffected by treatment with CCBs. hepatocytes do not express functional LVDCCs.
strongly implicate LVDCCs in NTBI accumulation into the myocardium during iron overload. Previous results have shown that the DCT-1/DMT-1 and transferrin transport systems are downregulated in their expression and activity in iron overload conditions. In contrast, one key estab- lished and unique feature of NTBI transport is the lack of downregula- tion in iron-overload tissues. Indeed, there was no difference in the density and voltage dependence (Fig. 4e) or kinetic properties (Fig. 4f) of LVDCC currents in cardiomyocytes from placebo-injected com- pared with iron-injected mice, suggesting that iron-mediated uptake through the LVDCC is not under a negative feedback control mecha- nism.
The DCT-1/DMT-1 and transferrin iron transport systems are regu- lated in a negative-feedback manner by both transcriptional (iron- responsive elements) and translational mechanisms3,29,31. In contrast, NTBI uptake into excitable cells such as cardiomyocytes is accelerated in iron-overload conditions27,28,30 and requires reduction of ferric iron (Fe3+) to ferrous iron (Fe2+) by a membrane-associated ferri- reductase system28–30,53,54. The heart and certain endocrine tissues (endocrine pancreas and anterior pituitary) are particularly suscepti- ble to secondary iron overload and possess a high abundance and activity of LVDCCs35,36,38,39. Because previous studies have shown that cardiac LVDCCs can transport Fe2+ ions in myocytes and Langendorff hearts32,33 and that cardiac LVDCC current increases in the presence of elevated reduced iron32, we hypothesized that NTBI uptake by the heart in vivo occurs mainly through LVDCCs in car- diomyocytes. Consistent with our hypothesis, myocardial iron deposi- tion is reduced, while survival, myocardial ultrastructure and cardiac function are improved, by treating iron-overloaded mice with amlodipine or verapamil at dosages yielding therapeutic plasma levels similar to those reported in patients treated with these drugs41,48–50. Moreover, peak plasma levels of CCBs in our mice inhibit the net car- diac LVDCC current per beat by more than 50%. The crucial role of LVDCCs in cardiac NTBI iron is further supported by our observation that iron injection in transgenic mice overexpressing functional LVDCC in cardiomyocytes35,51 resulted in greater myocardial iron accumulation and oxidative stress and deterioration in cardiac func- tion compared with iron-injected littermate control mice. Moreover, treatment of iron-overloaded transgenic mice with verapamil reduced myocardial iron levels by about 50% and markedly improved cardiac performance. In contrast to the heart, treatment with CCBs had no effect on iron levels in the liver, as expected from our hypothesis because this tissue is devoid of LVDCCs
In addition, unlike other candidate NTBI iron transporters such as DCT- 1/DMT-1 (refs. 29,31,34), no downregulation of LVDCC current was observed in myocytes from iron-overloaded mice. This lack of down- regulation of LVDCC current in iron overload, combined with the ability of elevated Fe2+to increased LVDCC current in cardiomy- ocytes32 is expected to increase cardiac iron transport, as required for the NTBI transport system, under iron-overload conditions
iron-mediated free-radical generation and elevated oxidative stress as major determinants of cardiotoxicity and apoptosis associated with iron-overload44,45,55, as well as in patients with pri- mary hemochromatosis46 and thalassemia47. Consistent with this sug- gestion, free-radical generation, as assessed by tissue aldehyde levels (the end-products of lipid peroxidation56,57), were markedly elevated in iron-overloaded mice and correlated strongly with iron levels as well as the structural and functional changes in the heart. Indeed, iron- overloaded mice showed elevated aldehyde and iron levels associated with impaired structure and function in the heart, all of which were exacerbated in iron-treated transgenic mice with elevated LVDCC current while being improved by CCB treatment.
Thoughts
My previous post discussed how Fe+2 could theoretically enter through something like a VGCC because of its same size and charge as Ca+2. I see this being a possibility in a deficient state where the body is unable to make tf, DMT1, and Frataxin. Or you have ROS/RNS or some other mechanism that damages these chaperones. In this situation you probably have reduced glutathione from a down regulation of the sulfation cycle which means the mitochondria and cell are prime for Fenton reactions. Also, GSH is needed to chaperone Cu+2 intracellularly which would add to the damage.
At this point does it really matter if it’s the Ca+2 or Fe+2 (etc.) causing the membrane permeability transition pore? The mitochondria, then cell, will die regardless. In a cancer cell this would be desired so I wonder if there is a way to leverage a situation like this? The problem seems to be like the glaring issue with chemotherapy, you would damage your healthy cells too. Fixing the higher order cause from deficiencies seems to be the more pertinent issue. We need SOD, GSH, catalase peroxidase, etc.
It is true that a Fe (and Cu) pool that becomes impossible to manage from people continually consuming foods high in these would cause a chaperone deficiency too. So it’s important to get things back in circulation.
Figures

Because of its redox properties, iron can catalyze the production of reactive oxygen species (ROS) that can be highly toxic (3). Therefore, under normal physiological conditions, iron is specifically transported in the blood by diferric transferrin (Tf) (4, 5). All tissues acquire iron by the binding of Tf to the transferrin receptor 1 (TfR1), with this complex then being in- ternalized by receptor-mediated endocy- tosis (4, 5) (Fig. 1A). Recent studies have demonstrated that the internalization of the Tf-containing endosome via the cyto- skeleton is under control of intracellular iron level
In vitro, iron release from Tf requires a “trap,” such as pyrophos- phate (13), but a physiological chelator serving this role has not been identified. In erythroid cells, once iron(III) is released from Tf in the endosome, it is thought to be reduced to iron(II) by a ferrireductase in the endosomal membrane known as the six-transmembrane epithelial antigen of the prostate 3 (14, 15) (Fig. 1A). After this step, iron(II) is then transported across the endosomal membrane by the divalent metal transporter-1 (DMT1) (16) and forms, as generally believed, the cytosolic low Mr labile or chelatable iron pool (17). This pool of iron is thought to supply the metal for storage in the cytosolic protein ferritin and for metabolic needs, including iron uptake by the mitochondrion for heme and ISC synthesis.
Iron can also be released from the cell by the transporter, ferroportin1 (18) (Fig. 1A). Ferroportin1 expression can be reg- ulated by the hormone of iron metabo- lism, hepcidin. Hepcidin is a key regulator of systemic iron metabolism (18) and is transported in the blood bound to α2- macroglobulin (18). Hepcidin secretion by the liver is stimulated by high iron levels and also inflammatory cytokines, such as interleukin-6 (19).
Once iron is trans- ported out of the endosome via DMT1 {divalent metal transporter 1}, it enters the chelatable or labile iron pool (Fig. 1A) that traditionally was thought to consist of low Mr complexes (e.g., iron-citrate) (17, 25, 26). The only strong evidence that such a pool exists comes from studies with che- lators that mobilize iron from cells (27, 28). However, it is just as likely that these com- pounds remove iron from organelles and proteins as it is that they chelate iron from genuine cytosolic low Mr complexes
Mitoferrin-1 is localized on the inner mitochondrial membrane and func- tions as an essential importer of iron for mitochondrial heme and ISC in erythro- blasts and is necessary for erythropoiesis (60). Mitoferrin-1 is highly expressed in erythroid cells and in low levels in other tissues, whereas mitoferrin-2 is ubiqui- tously expressed (61). The half-life of mitoferrin-1 (but not mitoferrin-2) in- creases in developing erythroid cells and this may be part of a regulatory mecha- nism aiding mitochondrial iron uptake
“kiss and run” hypothesis (11) (Fig. 1B). This model suggests that a direct transfer of iron from the Tf-containing endosome to the mitochondrion occurs, by-passing the cytosol
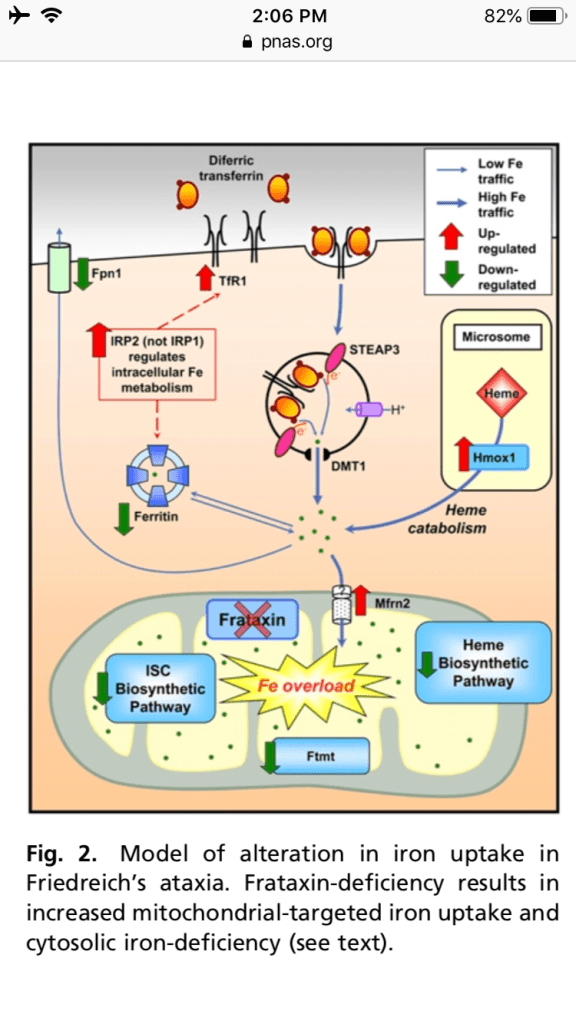
con- ditional frataxin-deletion in cardiomyocytes leads to depressed ISC and heme synthesis, mitochondrial iron-loading, TfR1 up-regu- lation, and thus increased iron uptake from Tf.
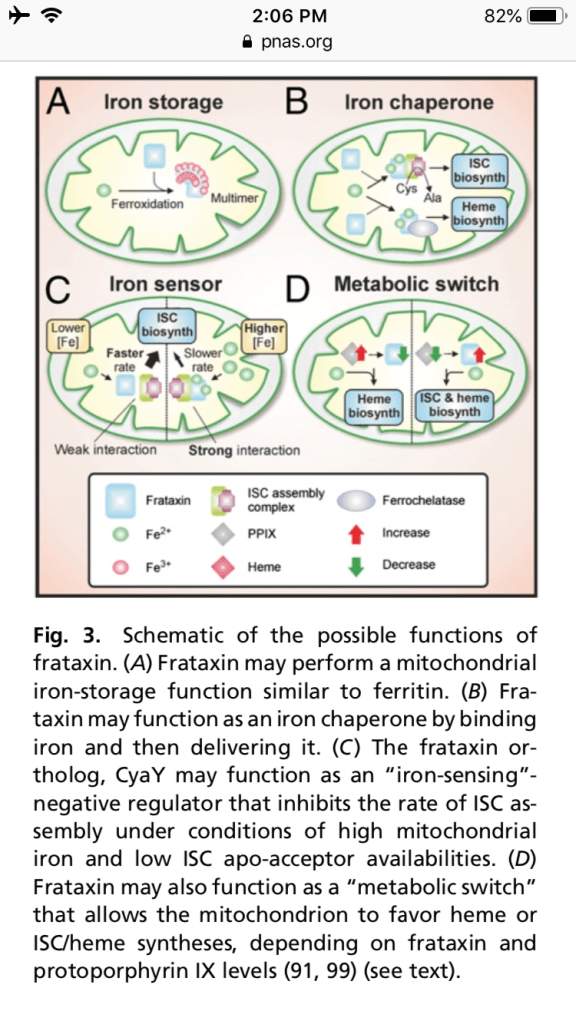
Frataxin has been proposed to function as a mitochondrial iron storage protein. Perhaps the most promising emerging role for frataxin is as an iron chaperone (Fig. 3B) for ISC and heme biosyntheses. Frataxin has been ob- served to interact with, and presumably donate iron to, iron-dependent proteins involved in ISC and heme biosyntheses.
may act as an “iron-sensor” (Fig. 3C) that negatively regulates the rate of ISC biosynthesis under conditions of high iron and low ISC apo-acceptor availabilities (89). If we extend this model to eukaryotic systems, a deficiency in fra- taxin expression is presumably deleteri- ous because the rate of ISC biosynthesis may exceed the availability of ISC apo- acceptors, resulting in the overproduction of ISCs that are unstable in an unbound form (89). Essentially, this model suggests that over and above functioning as an iron donor in ISC biosynthesis, frataxin may exert “kinetic control” over the rate of ISC biosynthesis, depending on the rela- tive availabilities of iron and ISC apo- acceptor
frataxin ex- pression is markedly decreased during Friend cell hemoglobinization (99), it
is possible that frataxin may be down- regulated during erythroid differentiation to allow higher rates of heme synthesis, potentially at the expense of decreased levels of ISC synthesis

Notes
Iron is needed not only for heme and iron sulfur cluster (ISC)-containing proteins involved in electron transport and oxidative phosphorylation, but also for a wide variety of cytoplasmic and nuclear functions, including DNA synthesis. the discovery of proteins involved in mitochondrial iron storage (mitochondrial ferritin) and transport (mitoferrin-1 and -2). In addition, recent work examining mitochondrial diseases (e.g., Friedreich’s ataxia) has established that communication exists between iron metabolism in the mitochondrion and the cytosol. This finding has revealed the ability of the mitochondrion to modulate whole-cell iron- processing to satisfy its own requirements for the crucial processes of heme and ISC synthesis. Knowledge of mitochondrial iron- processing pathways and the interaction between organelles and the cytosol could revolutionize the investigation of iron metabolism.
the mitochondrion is the sole site of heme synthesis and a major generator of ISCs, both of which are present in mitochondria and cytosol. Iron is an essential metal for the organism because of its unparalleled versatility as
a biological catalyst. Consequently, iron is a crucial element required for growth. However, the very chemical properties of iron that allow this versatility also create a paradoxical situation, making acquisition by the organism very difficult. Indeed, at pH 7.4 and physiological oxygen tension, the relatively soluble iron(II) is readily oxidized to iron(III), which upon hydrolysis forms insoluble ferric hydroxides. As a result of this virtual insolubility and po- tential toxicity because of redox activity, iron must be constantly chaperoned.
Still unknown how Fe crosses the outer mitochondria membrane.
Once iron is trans- ported into the mitochondrion it can then be used for heme synthesis, ISC synthesis, or stored in Ftmt. It is essential that mi- tochondrial iron is maintained in a safe form to prevent oxidative damage, as mi- tochondria are a major source of cytotoxic ROS (3). Hence, it is likely that as in the cytosol, iron is carefully transported within the mitochondrion in a form distal to the aqueous environment, deep in the hydro- phobic pockets of communicating proteins that form iron transport pathways.
Like a typical ferritin, Ftmt shows ferrox- idase activity and binds iron (66). Ftmt was found at the highest lev- els in the testes and the erythroblasts of sideroblastic anemia patients (67, 68). Mi- tochondrial ferritin has also been detected in the heart, brain, spinal cord, kidney, and pancreas (67). Unlike cytosolic ferritin, Ftmt is not highly expressed in the liver and spleen, suggesting that it plays a distinct role. These findings led to the hypothesis that Ftmt plays a role in protection from iron-dependent oxidative damage.
Being a major site of ISC assembly, the mitochondrion plays a pivotal role in the biosynthesis of ISC proteins (1, 2). ISCs consist of iron and sulfide anions (S2-), which assemble to form [2Fe-2S] or [4Fe-4S] clusters (2). These clusters form cofactors in proteins that perform vital functions, such as elec- tron transport, redox reactions, metabolic catalysis, and other such functions (70). In eukaryotic cells, more than 20 mole- cules facilitate maturation of ISC proteins in the mitochondria, cytosol, and nucleus (2). Functional defects in ISC proteins
or components involved in their biogenesis lead to human diseases (71, 72). Bio- synthesis of ISCs and their insertion into apo-proteins requires both the mitochon- drial and cytosolic machinery.
The mitochondrion contributes a yet-to-be discovered compound necessary for the biogenesis of ISCs outside of this com- partment [mitochondrial ISC assembly system] I.e., in the cytosol or other or- ganelles).
The third major mito- chondrial metabolic pathway that utilizes iron is that of heme synthesis that is ex- clusive to this organelle.
Apart from importing iron, the mitochondrion synthe- sizes heme and ISCs that subsequently are transported out to the cytosol. These export processes are important to understand, as decreased release of iron as heme or ISCs —or their precursors—can contribute to mitochondrial iron-loading.
It is unknown how heme is exported from the mitochondrion. However, its low solubility and highly toxic nature suggest an efficient heme-carrier must be involved. Considering this, heme-binding protein 1 has been identified as a candidate for this carrier (83). The expression of this mole- cule is ubiquitous, but it is also increased during erythroid differentiation and high levels are found in the liver (83). Heme- binding protein 1 binds one heme per mole of protein (83) and, although it could be a candidate for heme transport, direct evidence for this is lacking.
Fe+2 (also Co+2, Ni+2, Cu+2, Zn+2, and Sn+2) all have the same hydrated diameter as Ca+2 = 600pm. Given their same charge and hydrated diameter, I believe they could all go in through calcium channels.
This also shows why potassium is desired over Na, 300pm and 450pm respectively. And when the protoplasm structure changes Na will substitute for K (100pm vs 160pm unhydrated diameters). Dr Ling discusses this in terms of c-values. I’ll make a separate post about that later.
Paper
vitamin K plus C as a powerful redox-system, which forms a bypass between mitochondrial complexes II and III and thus prevents mitochondrial dysfunction, restores oxidative phosphorylation and aerobic glycolysis, modulates the redox-state of endogenous redox-pairs, eliminates the hypoxic environment of cancer cells and induces cell death. The analyzed data suggest that vitamin C&K can sensitize cancer cells to conventional chemotherapy, which allows achievement of a lower effective dose of the drug and minimizing the harmful side-effects.
developed large subcutaneous and intramuscular hemor- rhages. Henrik Dam has designated this factor as “Koagulation vitamin” (from Danish) or vitamin K in short, because of its vital role for normal haemostasis
Initial experiments with additions of lemon juice, cholesterol, cod-liver oil or ascorbic acid to the diet failed to prevent hemorrhages [1–3]. Later it has been discovered that vitamin K is an essential cofactor for post-translational modification of hepatic blood- coagulating proteins (as prothrombin, factors II, VII, IX and X) [3,4]. Vitamin K participates in the converting of their glutamic acid residues into γ-carboxyglutamic acid (Gla) residues.
role in bone metabolism, vascular calcification, regulation of cell growth and apoptosis
Vitamin K is a group of structurally related molecules that have a 2- methyl-1,4 naphthoquinone ring and a variable aliphatic chain (Fig. 1A). The variable aliphatic chain distinguishes two naturally oc- curring forms: vitamin K1 (phylloquinone) and vitamin K2 (menaqui- none). There is also a synthetic form of vitamin K without aliphatic chain (K3 or menadione), which is classified as a pro-vitamin. Vitamin K1 exists only in one phylloquinone structure, while vitamin K2 exists in multiple structures, which are distinguished by number of un-
saturated isoprenyl groups in the aliphatic chain [10].
Vitamin K1 is found in green leafy vegetables: broccoli, lettuce, spinach, fermented soy (natto), spring onions, cabbage, etc. The various forms of vitamin K2 (MK-4, MK-7, MK-10) are mainly synthesized by bacteria, especially in nutrient products as natto and yogurt [11,12].
Vitamin K2 is also found in meat, eggs, curd and cheese [7,10,13]. Vitamins K1 and K2 are transported by triglyceride-rich lipoprotein (TGRLP) to the liver, after intestinal absorption [13,14] (Fig. 1B). Vi- tamin K1 is metabolized and more than half the amount is excreted by the organism, while vitamin K2 is transported from the low density
lipoproteins (LDL) to extra-liver tissues [13,14]. Vitamin K2 is accu- mulated preferentially in peripheral tissues. High levels are detected in the brain, aorta, pancreas, fat and low levels are detected in the liver [15–17].
Pro-vitamin K3 is a synthetic lipid-soluble vitamin precursor, which can be converted to active vitamin K2 (menaquinone) after its alkyla- tion in the liver. In addition, pro-vitamin K3 could be metabolized to glucuronide and sulfate of reduced menadione, which are excreted in the urine [18–20]. Menadione has a relatively low toxicity and is ap- proved by the Food and Drug Administration for therapeutic purposes. It has been shown that pro-vitamin K3 directly affects the redox status of thiols (including thiol-containing compounds) and calcium home- ostasis [20]. The most intriguing property of menadione is its antic- ancer activity
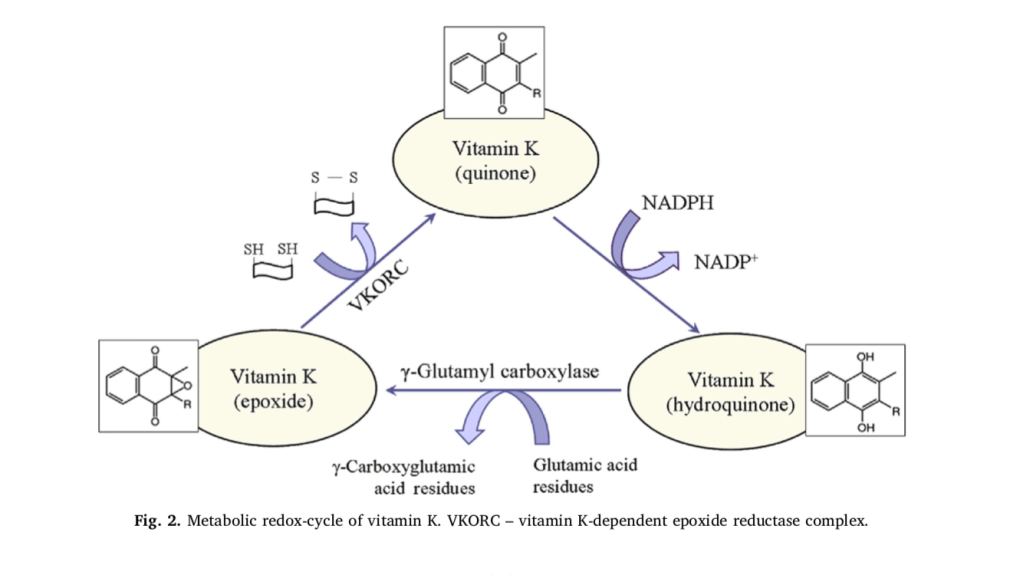
It is generally accepted that the essential role of vitamin K is related to the post-translational γ-carboxylation of glutamate residues of pro- teins (Fig. 2). This process occurs in the lumen of the endoplasmic re- ticulum and involves two enzymes: γ-glutamyl carboxylase and vitamin K-dependent epoxide reductase complex (VKORC). Both enzymes to- gether constitute the “vitamin K cycle”, which is a redox-cycle in its biochemical essence [21,22]. In nature, vitamin K exists in an oxidized form (quinone), but γ-carboxylation of glutamate residues requires its reduced form (hydroquinone). In the organism, quinone is reduced to hydroquinone by NAD(P)H and/or glutathione (GSH). In turn, hydro- quinone is converted to epoxide in the process of γ-carboxylation. The epoxide is converted to the initial oxidized quinone state by the VKORC, which is accompanied by a consumption of other reducing equivalents (mainly thiol-groups). Obviously, the activity of vitamin K may affect redox-homeostasis of cells and tissues and can be considered as a regulatory factor in redox-signaling. This function of vitamin K is very important due to variety of glutamic acid-rich proteins in the bones, arteries and soft tissues. For example, vitamin K influences the degree of carboxylation of osteocalcin – a small calcium-binding pro- tein, which is secreted by osteoblasts in bones and serves as an integral protein for the synthesis of bone matrix [23–25]. The biosynthesis of osteocalcin is regulated by hormones and growth factors, but its post- translational modification is regulated by vitamin K. Osteocalcin con- tains three Gla residues, which interact with calcium in the hydro- xyapatite-crystal lattice of bones [24]. γ-Carboxylation of glutamic acid residues of osteocalcin increases its affinity to calcium and respectively to hydroxyapatite [25]. It was shown, that decarboxylated osteocalcin cannot bind calcium, which emphasizes the essential role of vitamin K in the γ-carboxylation process [26,27]. In addition, vitamin K inhibits the activity of osteoclasts, thus preventing the breakdown of bones [27,28].
Vitamin K participates in the carboxylation of the matrix Gla-con- taining protein (MGP) of the arterial walls and plays a crucial role in maintaining their elasticity [7,8,29,30]. MGP belongs to the group of vitamin K2-dependent Gla-containing proteins. MGP is produced in bones and vascular smooth muscle cells and inhibits vascular calcifi- cation [30–32]. Its function is affected by inflammatory factors [32]. The importance of MGP for vascular homeostasis was demonstrated on MGP-deficient animals – all of them died of massive arterial calcifica- tion within 6–8 weeks after birth [5,33]. It was established that non- carboxylated MGP forms calcium depositions in the vascular walls [8,27,34]. The calcium phosphate crystals in the arterial wall directly attract macrophages and induce inflammation [27,35,36]. Studies have demonstrated that rats with vitamin K-deficiency and chronic kidney disease have an enhanced vascular calcification, which decreases after vitamin K supplementation [31,37]. Warfarin, an inhibitor of vitamin K reduction, affects γ-glutamyl carboxylation of MGP and induces vas- cular calcification in experimental animals [4,31,34,38]. The effect is abolished by vitamin K treatment [4,31,38]. These findings suggest that vitamin K-deficiency affects calcium homeostasis, which leads to vas- cular calcification and bone disorders.
Quinones can undergo one-electron reduction, producing inter- mediate semiquinone radicals, as well as two-electron reduction with production of hydroquinones (Fig. 3) [41]. Both reactions are accom- panied by consumption of superoxide radicals and reducing equivalents (as NADH, NADPH, glutathione), which are essential for cancer cell homeostasis [42,43]. It is generally accepted that superoxide is “on- cogenic ROS”, while hydrogen peroxide is “onco-suppressive ROS” [43–46]. Numerous studies suggest that cellular state, where the ratio tilts predominantly in favor of superoxide, inhibits apoptosis and pro- motes cell survival. If the ratio tilts in favor of hydrogen peroxide, this creates an intracellular environment suitable for induction of apoptosis and cell death.
this context, the decrease of superoxide due to vitamin K redox-cycle may explain, at least partially, its anticancer activity. On the other hand, semiquinone radical of vitamin K can convert transition metal ions, e.g. Fe3+ to Fe2+, thus inducing Fenton’s reactions and production of highly reactive and cytotoxic hydroxyl and hydroperoxyl radicals [10,49].
It is shown that pro-vitamin K3 induces oxidative stress in cancer cells via production of hydroxyl radicals and DNA strand-breaks.
Superoxide dismutase does not influence the anticancer effect of menadione [52,53]. However, antioxidant enzymes, involved in the depletion of “onco-suppressive” hydrogen peroxide (as catalase and glutathione peroxidase), decrease the anticancer effect of menadione [53,54]. Similar data have also been reported for transition metal chelators that suppress Fenton’s reactions and menadione-mediated cytotoxicity in cancer [55]. The described data suggest that pro-vitamin K3 leads to depletion of “oncogenic” superoxide and probably induces apoptosis via production of “onco-suppressive” hydroperoxides and cytotoxic hydroxyl radicals. This explains, at least partially, the antic- ancer activity of menadione. However, it should be noted that the in- duction of Fenton’s reactions and the production of hydroxyl radicals suggest for potential side-effects (cytotoxicity) of menadione on normal cells.
Other studies suggest that vitamin K induces apoptosis through different biochemical pathways, including alteration of intracellular calcium homeostasis and activation of the following pro-apoptotic factors: c-Jun N-terminal kinases (JNKs), Fas-dependent and Fas-in- dependent pathways, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [56–61].
Most of the vitamin K-induced pro- apoptotic factors are inflammatory signals, inducing overproduction of ROS (mainly hydroperoxides) and oxidative stress in cancer cells and tissues [43,48]. The data also suggest that vitamin K decreases the le- vels of superoxide, NAD(P)H and glutathione, which are essential for hypoxic behavior of cancer cells and their homeostasis.
glucose meta- bolism is a unique pathway. It can utilize oxygen if available (aerobic pathway – via conversion of pyruvate to acetyl-CoA) or it can function in the absence of oxygen (anaerobic pathway – via conversion of pyr- uvate to lactate). Both pathways are necessary for the functioning of cells, tissues and organs, but in norm, the aerobic pathway dominates the anaerobic. In cancer cells, the anaerobic oxidation of glucose is a major source of energy. In many cancers, the major cause of anaerobic glycolysis is mitochondrial dysfunction and inhibition of oxidative phosphorylation. Cancer cells are characterized by high levels of glu- cose, anaerobic oxidation of glucose (Warburg effect) and accumulation of lactate, compared to normal cells [43,44,48,79]. In last years, lactate dehydrogenase is receiving a great deal of attention as a potential di- agnostic marker or a predictive biomarker for many types of cancer and as a therapeutic target for development of new anticancer drugs [80–82].
severe defect in complex III of the mitochondrial electron transport chain (mitochon- drial dysfunction). Deficiency of reducible cytochrome b (cyt. b), which is accompanied by an effec- tive prevention of aerobic metabolism and oxidative phosphorylation. The authors bypass the deficient complex III by using pro-vitamin K3 and vitamin C as electron transfer mediators to carry the electrons from coenzyme Q (CoQ) to cyt. c. Thus, they succeeded to increase ATP production rate to about 65% of the normal state in mitochondria, as well as to decrease the lactic acidosis in the patient. The combination of vitamin C and K3 results in a production of more ATP, than in the case of vitamin C applied alone. The authors suggest that both vitamins can reduce directly cyt. c, since the reduction potential of cyt. c is over + 200 mV – more positive than those of menadione or ascorbate.
treatment of cells with vi- tamin C and K3 results in spikes in ATP production due to a shunt around a defective area of complex III of the mitochondrial electron- transport chain (between CoQ and cyt. c) [74,75,88–91]. This reaction causes a shift from anaerobic (glycolytic) to aerobic (oxidative) meta- bolism, which diminishes hypoxia and lactic acidosis.
case of dysfunc- tion in Complex-III (due to mutations in cyt. b), the electrons can not be transferred from CoQ to cyt. c. In the case of dysfunction in Complex-I (due to mutations in NADH dehydrogenase), the electrons are blocked on the first step of respiratory chain. It is well-known that both types of mutations are accompanied by superoxide production from complex-III and complex-I and suppression of ATP synthesis [43,48]. The oxidized vitamin K (KQ) can accept electrons from superoxide, converting con- sequently to semiquinone (KQH•) and hydroquinone (KQH2). Finally, the reduced vitamin K can transfer electrons directly to cyt. c. the oxidized vitamin C (dehydoroascorbate) can accept electrons di- rectly from Complex-I (NADH) or CoQ, converting consequently to semidehydroascorbate (Asc•) and ascorbate (Asc). Finally, the reduced vitamin C can transfer electrons to cyt. c. The two redox-cycles lead to a bypass between Complex-I (or Complex-III) and cyt. c. This will restore the oxidative phosphorylation and ATP synthesis. ATP, generated due to the vitamin C&K-mediated bypasses, prevents the anaerobic condi- tions, decreases the levels of “oncogenic” superoxide, decreases the levels of lactate, eliminates the lactic acidosis and hypoxia, and allows the cancer cells to trigger autophagia and “to kill themselves”.
redox cycles of vitamins C and K are cross-linked – ascorbate can reduce pro-vitamin K3, which is accompanied by a production of hydrogen peroxide (“onco-suppres- sive” and cytotoxic ROS) [92,93]. The reduction of vitamin K by vi- tamin C allows also a direct (superoxide-independent) reduction of cyt. c by vitamin K (KQH2). Silvera-Dorta et al. have also reported that vi- tamin C can reduce CoQ and the process is coupled with reduction of oxygen to hydrogen peroxide
The redox-system vitamin C&K3 could also influence the ratio be- tween the oxidized and reduced forms of the main endogenous redox- pairs: NAD+ /NADH, NADP+ /NADPH, GSH/GSSH, etc
The balance between oxidized and reduced forms is crucial for the cell behavior, as well as for cell survival or death. It is widely accepted that the increased mitochondrial oxi- dative stress and high levels of NAD(P)H and glutathione are distinctive features of the metabolic phenotype of cancer cells [43,44,47,48]. Since the reduction potentials of NAD(P)H, FADH2 and GSH are very low (below −200 mV), all substances can directly reduce vitamins C and K.
The redox-cycles of vitamins C and K are cross-linked and both vi- tamins can serve as associated redox-modulators (a redox-shuttle). The combination vitamin C&K can serve as a bypass between complex I (or complex-III) and cyt. c of mitochondrial respiratory chain and to donate electrons (directly or indirectly) to cyt. c, passing from reduced to oxidized form. Both vitamins can be also converted into reduced forms, consuming NAD(P)H and/or GSH. These processes are accompanied by an increase in ATP synthesis and accumulation of high levels of NAD(P) + and GSSH, which restores oxidative phosphorylation and aerobic glycolysis and eliminates the hypoxic environment. Тhe specific beha- vior of the cancer cells is disrupted and apoptosis is initiated. It is as- sumed that changing the ratio NAD(P)+/NAD(P)H and/or GSH/GSSH in cancer cells could be the “switch mechanism” from survival to cell death. Targeting defective mitochondria in cancer cells and preventing their dysfunction, as well as modulating redox-state of endogenous redox-pairs can be a successful strategy for anticancer therapy. This strategy allows target cytotoxicity, which is not directed to normal cells and tissues [43,48].
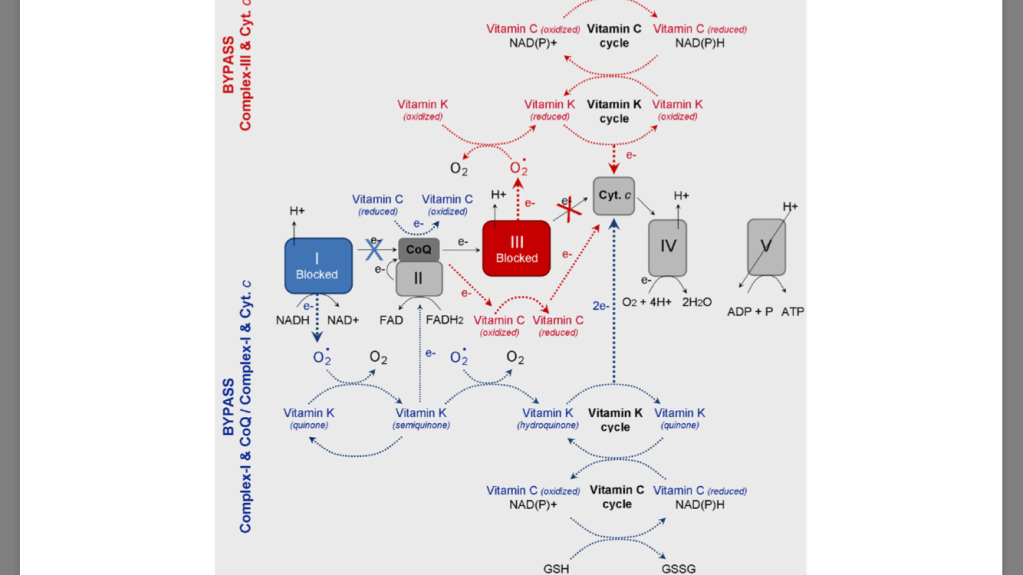

https://www.sciencedirect.com/science/article/pii/S2213231718300934
Thoughts
I wonder how this would change with the whole vitamin C complex? They mostly looked at the synthetic K3, I wonder what changes with K2? This consumes NAD and GSH so that needs to be addressed. Especially with the comments about the increased possibility of a Fenton reaction. If GSH is needed to transport Fe within the cell then taxing it further could make this more likely.
The part about hydrogen peroxide being onco-suppressive ROS is very interesting. And needing to tip the scale away from superoxide to hydrogen peroxide to stimulate apoptosis is very important. But again this needs reducing equivalents which require raw materials. Dr GRJ says glutathione comes indirectly from methylation. And glutathione reductase OSs FAD (B2) dependent which will fix the altered GSH:GSSG ratio. FAD = quinone, FADH2 = hydroquinone, and FADH = semiquinone. I wonder if FAD can do this too?
By passing complex I and III and seeing an increase in ATP by 65% is hugely important. I think finding more things that help bypass complexes staight to complex IV or the ATPase will be important to help many with low ATP states. Cold thermogenesis is known to stimulate thermogenin (UCP1) which transports protons from the inner mitochondrial space to the matrix, uncoupling the ETC. This occurs in brown adipose tissue. Do want more heat production or more ATP though?
Ling says we need ATP as a cardinal absorbent. “Cardinal adsorbents hold protoplasm in one or another state by a propagated polarization along the length of polypeptide chains. Adsorption or desorption of the cardinal adsorbents may alter the cooperative state of the protein- water-ion system in an “all-or-none” manner”. “proteins adsorbing a solute also polarize water; in this case the cardinal adsorbent controls at once the distribution of adsorbed solute and the free solute dissolved in the water. In nature, perhaps the more frequently observed case is one in which different proteins are involved in solute adsorp- tion and in water polarization”. But you still need the “heat source” for the jello. So maybe the UCP making heat is important.
Then you have Dr Arturo Herrera whose research shows melanin splits the water in the cell making energy. I wonder what frequency of energy? I bet it’s IR so the sunlight provides the heat source during the day and the reason our body temperature drops at night is to stimulate the UCP to allow the mitochondria to release heat (IR). Herrera says the melanin is around the cell nucleus. I wonder if you would be able to see the difference? The heat source coming from one relatively larger nucleus during the day and from many smaller mitochondria at night.
News
I love that Thomas Huxley, a biologist, wrote the opening to the first Nature. He is the grandfather of Andrew Huxley; also a biologist of the Hodgkin-Huxley conductance-based model fame. T Huxley’s use of Goethe is interesting, given how Goethe seems to be talking about God but attributing His energies to mother nature. T Huxley was agnostic so he stopped at empirical data.
A foreign agency, [insect], is required for plants to cross-fertilize. Winter plants are able to self-fertilize by changing their structure within or after the flower has opened. Plants can cross-fertilize to others close to them of the same or different species. {nearest neighbor interactions?} Abruptly bringing plants indoors (warmer temperature), will cause them to discharge their pollen faster.
A comedic piece discussing a reverends dismissal and argument against T Huxley’s views. And the editorial seems to be an early argument that scientists work for mankind and not only for themselves or their fellows. {This is obviously a fallacy as man is fallen} Science shouldn’t be enforced on others. However, data and observation should be shared and discussed. “It is impossible to separate science from other knowledge and from daily life”. {this is why established views take a generation passing to begin to be challenged}
Solar eclipses stop the glare caused by he reflection of sunlight on earths atmosphere. This allows anything surrounding the sun that is normally inhibited by the glare to be seen. This allows one to see the pearly white corona and red prominences {solar flares?}. The spectroscope was used to study the “red flames” and showed mostly H. There was major disagreement about the source of the corona effect. The solar spectrum shows the Fraunhofer lines of H, Na, Fe, and Mg.
Books Received
Many stone implements were used in the Bronze Age.
Look up Hansen’s method for calculating planet disturbances; planet Pomona; Themis {they seem to be moons of Jupiter from the context clues}; bouvards tables for Jupiter and Saturn. The planets effect each other {if only modern day science would allow for this}
News
Teaching science to children: experimental mechanics, chemistry, and physiology. Also systemic botany to teach observation. Electricity interests boys and should be fostered. “Boys make nothing their own so thoroughly as that which they select themselves”. So let them branch out from these topics. At least 3h/wk -> 2y mechanics + 2y chemistry + 1y botany + what remains for physiology. Start when 11yo. Year 1: tons of experiments and some easy problems; reproduce lectures and drawings by hand each week. Year 2: master a good book. Year 3: lectures in inorganic chemistry with a textbook and experiments shown. Year 4: all laboratory. Year 5: botany, henslows schedules – will know classification and be able to identify local flora and fauna. Remaining time: physiology first human then comparative (Huxley’s book).
Mechanics: air pump, pulleys, models of force pump, common pump, Keith Johnston’s scientific maps, newths natural philosophy second year.
Take daily walks and understand engines at work.
Chemistry: still, stove, has jars, pneumatic trough, tubing, chemicals, test tubes, wash bottle, 24 test solutions, blow pipe, beakers, Roscoes or Williamson’s textbook.
Botany: professor oliver lessons, Lindleys descriptive botany
Meteorogical instruments and a telescope.
“Culture and refinement brings fewer pupils up to a given mark within a given time; it what he has taught remains with them; they never forget or fall back”
Habit and desire of intellectual improvement is what school is supposed to provide. Must make learning pleasurable for students to learn.
The amount of water mixed with phosphoric acid dictates what compounds are made.
The slow oxidation of phosphorous by atmospheric air emits light.
Water of crystallization = water + various salts
Chemistry
Uranium was found to be one of the densest metals
Physiology
Cholera results from a cholera poison (z) and then there is a cholera germ (x) which “of itself will not produce cholera symptoms. {bechamps terrain theory proven correct again… the bacteria shows up to deal with the poison}. “It may remain, and probably may multiply in the human body, and be carried in or on the body from place to place without of itself producing cholera. Cholera symptoms can only be brought about by z, and x can only give rise to cholera, indirectly, by generating z. But x, in order that it may generate z, must come into contact and act on another substance (y). So x germinates into z only through meeting with y. So x is like dad and y like mom who make z which is sterile or can only produce x. With z you have some x which clings to the products of z.
Societies and academies
The last light from the sun when setting is bluish-green
The moon gives off heat and the amount increases rapidly to the full moon.
Yellow and red rays cause faster evaporation and decomposition of carbonic acid than blue and violet.
